Traumatic Spinal Cord Injury - Overview Of Its Mechanism And Modeling Approach
Traumatic spinal cord injury is a crippling neurological disorder that has a significant financial effect on afflicted people and the healthcare system.
Author:Karan EmeryReviewer:Katharine TateJan 09, 202379 Shares1.7K Views
Traumatic spinal cord injuryis a crippling neurological disorder that has a significant financial effect on afflicted people and the healthcare system.
According to this articlefrom Spinal-Injury.net, a person can suffer from spinal trauma if they sustain an injury that causes damage to their spinal cord, spinal column, or the bones that surround their spinal cord. Damage to the spinal cord, which includes the nerves that convey information between the brain and the rest of the body, can produce permanent changes in the functions of the body.
In North America, 12,500 new incidences of spinal cord injury are reported each year.
More than 90% of spinal cord injury instances are traumatic and result from traffic accidents, violence, sports, or falls.
Spinal cord injury has a documented male-to-female ratio of 2:1, and it occurs more commonly in adults than children.
The age distribution is bimodal, with a first peak of young people and the second high of those over 60.
The damage and retained functions significantly impact the life expectancy of traumatic spinal cord injury patients.

Spinal Cord Injury - diagnosis, treatment, recovery.
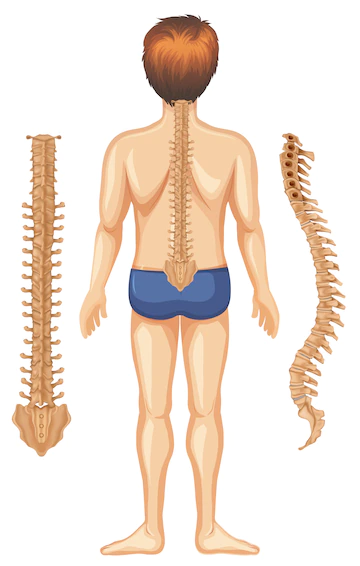
Types Of Spinal Injuries
Cervical, thoracic, lumbar, and sacral spinal cord injuries are the four types.
The spinal cord is encircled by bone rings known as vertebrae. The spinal column is made up of these bones.
The higher the lesion occurs in the spinal column, the more dysfunction a person would feel. The vertebrae are called from their placement.
The cervical vertebrae are the seven vertebrae in the neck. C-1 is the top vertebra, C-2 is the next, and so on.
Injury to the cervical spinal cord generally results in loss of function in the arms and legs, leading in quadriplegia and spinal cord paralysis.
The thoracic vertebrae are the 12 vertebrae in the thorax. T-1 is the first thoracic vertebra to which the top rib connects.
Thoracic spinal cord injuries often affect the chest and legs, resulting in paraplegia. The lumbar vertebra is the vertebra in the lower back between the thoracic vertebra, where the ribs join, and the pelvis (hip bone).
The sacral vertebrae are located from the pelvis to the end of the spinal column. Injuries to the five lumbar vertebrae (L-1 through L-5) and the five sacral vertebrae (S-1 through S-5) often result in some loss of function in the hips and legs.
Mechanism Of Traumatic Spinal Cord Injury
The secondary injury develops within minutes of the first injury and may linger for weeks or months, causing progressive damage to the spinal cord tissue around the lesion site.
It was observed during a study on spinal cord damage in dogs that eliminating the post-traumatic hematomyelia improved neurological prognosis.
More spinal cord damage is caused by the presence of "biochemical components" in the necrotic hemorrhagic lesion.
Secondary injury is still used in the field to define cellular, molecular, and biochemical activities that continue to self-destruct spinal cord tissue and impede neurological recovery after spinal cord damage.
There are three types of secondary injury: acute, subacute, and chronic.
The initial phase of spinal cord injury includes vascular damage, ionic imbalance, neurotransmitter accumulation (excitotoxicity), free radical generation, calcium influx, lipid peroxidation, inflammation, edema, and necrotic cell death: apoptosis, demyelination of surviving axons.
The acute secondary damage components listed below contribute to the pathophysiology of spinal cord injury.
Hypoxia And Ischemia
One of the early outcomes of initial damage is disruption of spinal cord arterial supply and hypo-perfusion.
Hypovolemia and hemodynamic shock caused by severe bleeding and neurogenic shock limit spinal cord perfusion and ischemia in spinal cord injury patients.
Larger arteries, such as the anterior spinal artery, usually stay intact, but rupture of smaller intramedullary vessels and capillaries sensitive to severe injury results in leukocyte and red blood cell extravasation.
Blood flow to the spinal cord is further disrupted by increased tissue pressure in the edematous damaged spinal cord and hemorrhage-induced vasospasm in intact arteries.
In rat and monkey models of spinal cord injury, blood flow at the lesion epicenter decreases gradually during the first several hours after damage.
It stays low for up to 24 hours.
Gray matter is more vulnerable to ischemia injury than white matter because it comprises neurons with high metabolic requirements and has a 5-fold greater density of capillary beds.
After the damage, white matter blood flow often recovers to normal within 15 minutes; however, the gray matter has many hemorrhages and, consequently, re-perfusion usually does not occur for the first 24 hours.
Vascular injury, bleeding, and ischemia eventually result in cell death and tissue damage through various processes, including oxygen deprivation, ATP loss, excitotoxicity, ionic imbalance, free radical production, and necrotic cell death.
Excitotoxicity, Ionic Imbalance, And Oxidative Damage
Glutamate is the principal excitatory neurotransmitter in the central nervous system (CNS).
Glutamate binds to ionotropic (NMDA, AMPA, and Kainate receptors) and metabotropic receptors, resulting in calcium influx inside cells.
When intracellular Ca2+ levels grow, astrocytes may also expel excess glutamate extracellularly. Mitochondria have an essential role in calcium-dependent neuronal death.
During glutamate-induced excitotoxicity, NMDA receptor overactivity promotes mitochondrial calcium overflow in neurons. Ca2+ is transported into the mitochondria via the mitochondrial calcium uniporter (MCU).
Shortly after spinal cord damage, an influx of Ca2+ activates many protein kinases and phospholipases.
Riluzole, a Na+ channel blocker, lowers tissue damage and improves functional recovery in spinal cord injury, emphasizing the importance of sodium in secondary injury processes.
An increase in Na+ concentration activates the Na+/H+ exchanger, increasing intracellular H+. Lipid and protein oxidation is one of the critical secondary damage mechanisms after spinal cord injury.
Lipid and protein oxidation after spinal cord damage has several severe biological consequences.
These include mitochondrial respiratory and metabolic failure and DNA alterations leading to cell death.
Cell Death
Cell death is a significant event in the secondary damage processes that affect neurons and glia after spinal cord injury.
Necrosis and apoptosis were the first two main cell death mechanisms identified.
Necroptosis and autophagy are two novel kinds of cell death discovered in recent research.
Receptors control the process of necroptosis. It is triggered by TNF receptor 1 (TNFR1) and requires RIPK1 and RIPK3.
In rats, apoptosis begins as early as 4 hours after injury and peaks at 7 days. Injury-induced Ca2+ influx is the primary cause of apoptosis, which activates caspases and calpain, enzymes involved in protein breakdown.
Human post-mortem studies revealed that Fas-mediated cell death is associated with oligodendrocyte apoptosis and the inflammatory response after spinal cord damage.
Dysregulation of autophagy results in neuronal death, and blocking it has been related to neurodegenerative diseases such as Parkinson's and Alzheimer's.
Autophagy aids cell survival by eliminating toxic proteins and mitochondrial damage.
Immune Response
Neuroinflammationis a secondary injury process that occurs both locally and systemically.
Inflammation was thought to aggravate spinal cord damage.
Inflammation following spinal cord damage may be beneficial or detrimental depending on the time and immune cell activation.
Microglia/macrophages/microglia activation and autoantibodies against spinal cord antigens are caused by injury.
Astrocytes use IL-1R1-Myd88 to recruit neutrophils. TGF and IL-10 are produced by astrocytes, which create a pro-regenerative M2-like phenotype in microglia/macrophages.
Blood neutrophils infiltrate the spinal cord within hours after injury. Their removal makes healing and rehabilitation more difficult.
Cytokines, chemokines, and growth factors are all affected by the neutrophil reduction.
Microglia/macrophages populate the injured spinal cord within 2-3 days after injury.
Blood monocytes and bone marrow myeloid progeny give rise to macrophages.
T and B cells are essential for immune healing after spinal cord damage.
Autoreactive T lymphocytes kill neurons and glial cells. T cells' production of pro-inflammatory cytokines and chemokines influences brain cell activity and survival.
In individuals with spinal cord damage and multiple sclerosis, myelin-specific proteins like MBP activate circulating T cells.
Spinal cord damage inhibits B-cell activation and antibody production. Immunomodulatory Breg B cells aid in the healing of the spinal cord.
Cardiovascular, renal, and reproductive dysfunctions post-spinal cord injury are exacerbated by autoreactive immune cells.
Scar Tissue In The Glia
Glial scarring is a multifactorial phenomenon generated by several populations in the injured spinal cord.
Both resident and invading inflammatory cells have a role in glial activation and scar formation.
Fibroblasts, pericytes, and ependymal cells, as well as glial and immune cells, contribute to its formation.
Activated astrocytes play an essential role in the formation of the glial scar. Blood-brain-barrier regeneration also requires reactive astrogliosis.
Leukocyte infiltration, cell death, myelin breakdown, and functional recovery are all impeded by blocking this pathway.
Chondroitin sulfate proteoglycans (CSPGs) are well known for inhibiting axonal regeneration by the glial scar.
CSPGs are substantially elevated after injury and reach a peak of expression two weeks later. After spinal cord damage, the inhibitory effects of the astrocytic glial scar on axonal regeneration have recently been called into doubt.
Sofroniew and colleagues revealed that spontaneous axon regrowth did not occur after scar ablation or prevention using various transgenic mouse models.
Experimental Models For Spinal Cord Injury
In recent years, there has been a rise in interest in employing non-human primates and other larger animals as intermediate pre-clinical models.
Because of their size, they are more suited for drug development, bioengineering breakthroughs, electrophysiology, and rehabilitative research.
Models Of Transection
An entire transection spinal cord damage model is relatively easy to recreate. It is, however, less relevant to human spinal cord damage.
Transection models are suitable for studying axonal regeneration and developing biomaterial scaffolds to bridge the gap between a damaged spinal cord's proximal and distal regions.
These models are less useful in generating and testing new therapies since they result in less severe injury and higher amplitude of spontaneous repair.
Models Of Persuasion
In 1992, Gruner established the impactor paradigm at New York University.
The original New York University impactor consisted of a metal rod with a fixed weight (10 g) that could be dropped from a certain height onto the exposed spinal cord to produce damage.
The most recent version, Multicenter Animal Spinal Cord Injury Study III, was launched in 2012 and included both electromagnetic control and digital recording of impact parameters.
The Multicenter Animal Spinal Cord Damage Study, Infinite Horizon, and Ohio State University impactor devices were used to produce spinal cord injury in rats efficiently.
Multicenter Animal Spinal Cord Injury Study is a computer-controlled electromagnetic impactor invented in 1987 and improved in 1992.
The Infinite Horizon impactors have the potential to cause mild, moderate, or severe spinal cord damage (for example, 100, 150, and 200 kdyn).
The irregularity of the clamps on these impactors in firmly maintaining the spinal column during the impact may result in inconsistent parenchymal damage and neurological deficits.
Models Of Compression
Clip compression is the most often used compression model of spinal cord damage in rats and mice.
In this model, a tailored aneurism clip with a calibrated closure force is applied to the spinal cord for a set time (usually 1 min).
The level of injury may be calibrated and adjusted by varying the power of the clip and the period of compression.
For example, a 50 g clip for 1 minute frequently causes significant spinal cord damage. Calibrated forceps compression has also been employed in animals to cause spinal cord damage.
This model uses calibrated forceps with a spacer to compress the spinal cord bilaterally.
This model misses the initial impact and chronic damage found in most genuine traumatic spinal cord injury patients.
In general, all compression models have the same limitation: the velocity and amount of force are unmeasurable.
People Also Ask
What Causes A Traumatic Spinal Cord Injury?
Road traffic accidents fall, and violence is the primary cause of spinal cord injury. While work or sports-related injuries also account for a significant fraction of traumatic spinal cord injury.
Can You Fully Recover From A Spinal Cord Injury?
Unfortunately, there is no method to repair spinal cord injury. However, researchers are constantly developing novel therapies, including prostheses and drugs, that may accelerate nerve cell regeneration or enhance the function of the nerves that survive a spinal cord injury.
What Is The Most Serious Permanent Effect Of Spinal Cord Trauma?
A spinal cord injury is one of a person's most devastating ailments. This form of physical damage may cause paraplegia, the inability to move the lower body, quadriplegia, or the inability to move the body.
What Is The Best Treatment For Spinal Cord Injury?
Following rehabilitation practices may assist people with SCI, including:
- Muscle strengthening, communication, and mobility physical therapy
- Wheelchairs, walkers, and leg braces are examples of assistive aids.
- Communication with adaptive devices
- Fine motor abilities were the emphasis of occupational therapy.
Conclusion
Traumatic spinal cord injury has diverse and complicated pathogenesis.
While pre-clinical research on spinal cord injury has continued for over a century, our knowledge of spinal cord injury processes has grown significantly in the last few decades.
This is mainly owing to the development of novel transgenic and preclinical animal models, which have aided in the quick discovery of spinal cord injury mechanisms.
Although spinal cord injury research has made significant progress, more effort has to be made to transfer findings from animal studies to therapeutic applications in people.

Karan Emery
Author

Katharine Tate
Reviewer
Latest Articles
Popular Articles